В случае, если у нашей компании есть своя система фармаконадзора, как мы будем передавать Вам ведение фармаконадзора на аутсорсинг? Как будет происходить передача дел без приостановления ведения фармаконадзора?
АНО «ННЦФ» имеет большой опыт работы как с компаниями, которые только выходят на рынок, так и с компаниями, у которых уже выстроена система фармаконадзора. При заключении Договора сотрудники АНО «ННЦФ» готовят карту передачи дел, в которой четко указываются сроки передачи, а также назначение. Согласно данной карте в течение месяца Ваша компания полностью перейдет в руки АНО «ННЦФ».
Предоставляете ли вы план работ по подготовки досье для получения РКИ?
Да, сотрудники АНО «ННЦФ» проводят аудит Ваших документов и, согласно аудиторскому отчету, предоставляют Вам наиболее оптимальную стратегию регистрации именно для Вас, в которой четко указаны этапы и сроки для получения РКИ.
Берете ли вы срочные заказы с ограничением во времени по пользовательскому тестированию?
Да, АНО «ННЦФ» имеет достаточно большую базу респондентов, и может провести пользовательское тестирование в сроки до 7 рабочих дней.
Каков порядок действий при заказе услуги у Вас? Что от нас требуется?
При заключении с Вами Договора, сотрудник отдела продаж закрепляет за Вами ответственного менеджера, который будет сопровождать Вас на протяжении всего Договора. В течение 2 рабочих дней после получения от Вас скана, менеджер запросит необходимые материалы для работы.
Возможно ли проведение аудита онлайн?
Да, конечно. После заключения Договора предварительно направляется план проведения аудита, согласовываются с Вам даты аудита, а также формат ( очно/заочно). А по завершению аудита направляется аудиторский отчет, который с Вами согласовывается.
Необходимо ли держателю регистрационного удостоверения организовывать систему фармаконадзора в случае, если он не является производителем лекарственного препарата?
Да, согласно требованиям Правил надлежащей практики фармаконадзора GVP ЕАЭС ответственным за организацию системы фармаконадзора является держатель регистрационного удостоверения.
Возможно ли начать создание системы фармаконадзора в организации только после производства и выпуска в гражданский оборот первой серии лекарственного препарата?
Система фармаконадзора должна быть организована на протяжении всего жизненного цикла обращения лекарственного препарата, включая мониторинг нежелательных явлений на этапе проведения клинических исследований. Кроме того, на этапе проведения регистрационных действий в уполномоченный орган предоставляется информация об уполномоченном лице по фармаконадзору.
Какие существуют риски самостоятельного ведения системы фармаконадзора в организации?
Потенциальными рисками для держателя регистрационного удостоверения являются дефицит квалифицированных сотрудников в области фармаконадзора, необходимость в приобретении и обслуживании компьютеризированных валидированных систем, отслеживания и соблюдения национальных регуляторных требований в других странах обращения лекарственных препаратов.
Достаточно ли внесения информации об уполномоченном лице по фармаконадзору в составе регистрационного досье?
Нет, информация о назначении уполномоченного лица по фармаконадзору должна быть направлена в Федеральную службу надзору в сфере здравоохранения Российской Федерации.
Какие данные об уполномоченном лице по фармаконадзору включаются в состав мастер файла по фармаконадзору?
Документы, подтверждающие профильное образование, наличие квалификации и опыта организации системы фармаконадзора.
Требуется ли дополнительное назначение контактного лица по фармаконадзору на национальном уровне (КЛФ) в стране проживания уполномоченного лица по фармаконадзору?
Нет, дополнительное назначение контактного лица по фармаконадзору на национальном уровне в данном случае не требуется.
Каким образом должно быть организовано взаимодействие между уполномоченным и контактным лицом по фармаконадзору?
Рекомендуется разработка и включения данных вопросов в стандартные процедуры/ стандарты/ положения и другие соответствующие документы, где включается информация о сроках, каналах и форматах передачи информации и формы отчетности.
С какой периодичностью необходимо подавать в уполномоченный орган периодический отчет по безопасности лекарственного препарата (ПООБ), если лекарственный препарат отсутствует в утвержденном перечне подачи ПООБ регуляторного органа?
Если в утвержденном перечне нет необходимого лекарственного препарата, то подача ПООБ осуществляется в соответствии с требованиями надлежащей практики фармаконадзора ЕАЭС (каждые 6 месяцев от международной даты регистрации в течение первых 2 лет; ежегодно в течение последующих 2 лет; далее - каждые 3 года)
Возможна ли подача периодического отчета по безопасности в регуляторный орган на английском языке?
Направление периодического отчета по безопасности (ПООБ) в регуляторный орган осуществляется в формах:
- документа, адаптированного под требования ЕАЭС и переведенного
на русский язык;
- ПООБ на английском языке с переводом основных разделов на
русский язык
Требуется ли регламентация взаимоотношений с контрактными производственными площадками в части сбора информации о нежелательных реакциях?
Согласно требованиям надлежащей практики фармаконадзора GVP ЕЭАС держатель регистрационного удостоверения должен внедрить и применять все механизмы и источники сбора полной информации о случаях подозреваемых нежелательных реакций. Контрактные производственные площадки и дистрибьюторы также являются источниками получения данных сообщений, поэтому наличие соглашения об обмене данными по безопасности будет обеспечивать выполнения регуляторных требований по фармаконадзору.
Требуется ли повторная подача информации о нежелательной реакции в регуляторный орган, если данная информация была подана ранее медицинской организацией?
Нет, в этом случае повторную подачу информации в регуляторные органы осуществлять не нужно.
Какие препараты относятся к воспроизведенным?
Воспроизведенный лекарственный препарат («дженерик») - лекарственный препарат, который имеет такой же количественный и качественный состав действующих веществ и ту же лекарственную форму, что и оригинальный препарат, и биоэквивалентность которого оригинальному лекарственному препарату подтверждается соответствующими исследованиями биодоступности. Различные соли, эфиры, изомеры, смеси изомеров, комплексы или производные действующего вещества признаются одним и тем же действующим веществом, если их безопасность и эффективность существенно не отличаются. Различные лекарственные формы для приема внутрь с немедленным высвобождением признаются в рамках исследований биодоступности одной и той же лекарственной формой.
Какое государство являются референтным и государством признания?
"Референтное государство" - государство-член, осуществляющее подготовку экспертного отчета об оценке безопасности, эффективности и качества лекарственного препарата на основании экспертизы лекарственного препарата в соответствии с настоящими Правилами. "Государство признания" - государство-член, в котором лекарственный препарат зарегистрирован (регистрируется) с проведением экспертизы, включающей оценку экспертного отчета об оценке безопасности, эффективности и качества лекарственного препарата, подготовленного референтным государством.
В каких случаях применяется упрощенная процедура приведения регистрационного досье в соответствие с требованиями ЕАЭС?
Упрощенная процедура применяется только для лекарственных препаратов, зарегистрированных в заявляемых государствах - членах ЕАЭС (без расширения географии).
Представление каких документов Модуля 3 Регистрационного досье в рамках упрощенной процедуры приведения в соответствие с требованиями Союза не являются обязательными?
В рамках проведения процедуры приведения в соответствие с требованиями Союза в составе модуля 3 регистрационного досье представление документов, указанных в пунктах 3.2.S.2.3 - 3.2.S.2.6. 3.2.S.4.3, 3.2.S.4.5, 3.2.S.7.2, 3.2.P.2.2.3, 3.2.P.2.3-3.2.P.2.6, 3.2.P.4.3, 3.2.P.4.6, 3.2.A. 3.2.А.2, 3.2.А.3, 3.2.А.3.3, и 3.2.А.3.7 настоящих требований, не является обязательным (за исключением регистрационного досье биологических лекарственных препаратов). В случае их непредставления в рамках указанной процедуры документы необходимо представить в рамках подтверждения регистрации или при внесении изменений в регистрационное досье.
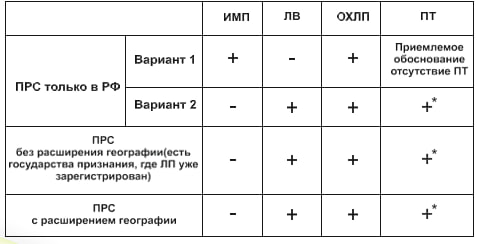
Возможно ли внесение изменений в регистрационное досье одновременно с приведением досье с требованиями Союза?
При приведении в соответствие с требованиями Союза, заявителем могут одновременно вноситься изменения в регистрационное досье зарегистрированного лекарственного препарата. В этом случае процедура внесения таких изменений и оценка досье на соответствие актам, входящим в право Союза, осуществляется в соответствии с Приложением N 19 к Правил регистрации
Чем обусловлена необходимость проведения валидационного пилотного этапа пользовательского тестирования?
Пилотный этап пользовательского тестирования необходимо проводить с целью проверки корректности и понимания формулировок задаваемых вопросов респондентам и снижения рисков при проведении основного исследования.
Что делать, если необходимо провести пользовательское тестирование, но нет готового сверстанного макета?
Национальный научный центр фармаконадзора предоставляет такую услугу как «верстка макета» с учетом желаемых характеристик.
Какие материалы потребуются для проведения пользовательского тестирования?
Для проведения пользовательского тестирования потребуются проект листка-вкладыша и макет листка-вкладыша к лекарственному препарату.
Что делать, если в ходе пользовательского тестирования получены неудовлетворительные результаты?
В случае получения неудовлетворительных результатов пользовательского тестирования предполагается внесение изменений в содержательную часть листка-вкладыша и/или макет листка-вкладыша. После получению обновленной версии листка-вкладыша этап пользовательского тестирования повторяется.
Из каких этапов состоит проведение пользовательского тестирования в Национальном научном центре фармаконадзора?
1 этап: Разработка и согласование с заказчиком протокола
пользовательского тестирования листка-вкладыша, включая разработку
опросника;
2 этап: Проведение интервью;
3 этап: Разработка отчета и резюме пользовательского тестирования на основании полученных результатов.
Где проводится пользовательское тестирование?
Пользовательское тестирование проводится в отдельных кабинетах, предназначенных для проведения интервью, где респондентам предоставляются необходимые комфортные условия для прохождения опроса.
В течение какого времени проводится набор участников опроса для пользовательского тестирования?
Зависит от наличия базы респондентов. В Национальном научном центре база потенциальных участников насчитывает численность более 4 тысяч, что позволяет набрать и провести исследование в течение 1 недели.
Какая процедура заполнения опросников пользовательского тестирования?
Данные в опросник вносятся интервьюером разборчиво черной шариковой ручкой для возможности получения четких копий страниц со слов респондента (дословно), параллельно фиксируя время начала и завершения интервью, а также комментарии респондента касательно макета листка-вкладыша при их наличии. Процесс интервью проходит под камерами видеонаблюдения.
Возможно ли проведение фокус-группы в части пользовательского тестирования?
Да, чаще всего в фокус-группе возникает необходимость при актуализации листка-вкладыша после получения запроса эксперта и проводится на определенном разделе листка-вкладыша, в который вносились изменения.
Гарантирует ли положительный результат проведения исследований биоэквивалентности лекарственного препарата проведение предварительного теста сравнительной кинетики растворения in vitro (TCKP)?
Нет. ТСКР является прогностическим инструментом на ранних этапах разработки препарата, однако не позволяет доподлинно предопределить биодоступность препарата в организме человека и, следовательно, eго биоэквивалентность.
Есть ли требования к максимальному сроку хранения биообразцов?
Максимальный срок хранения биообразцов определяется опытным путем и зависит от природы аналита. Нормативной базой максимальные сроки хранения биообразцов не регламентируются. Как правило, срок хранения биообразцов при температуре -20°С не должен превышать 1 месяца, в то время как хранение при -70°С обеспечивает сохранность до года и более.
Какой объем исследований может быть проведен для гибридного препарата в инъекционной форме? (оригинатор - таблетки)
Так как в исследовании новый путь введения и, по всей видимости, новые дозировки, то клиническое исследование должно быть спланировано следующим образом: I фаза - проведение фармакокинетического анализа и подтверждение переносимости нового пути введения, IIb фаза подтверждение дозировок, III - подтверждение эффективности. Данное разделение по фазам является условным, и программа может включать вместо исследований IIb и III фазы - одно объединенное исследование фазы II-III.
Как планируются исследования биоэквивалентности, когда нет опубликованных результатов ранее проведенных исследований? На чем основывается выбор дизайна и размера выборки?
В данном случае планируется исследование с адаптивным дизайном, которое проводится в два этапа. На первом этапе оценивается БЭ на ограниченной выборке субъектов исследования, и если БЭ не подтверждается, то проводится второй этап исследования на дополнительной выборке субъектов, размер которой определяется по данным первого этапа.
Как планируется дизайн исследований III фазы в случае невозможности выполнения фармакокинетического исследования биоэквивалентности?
Как правило, это сравнительные исследования с гипотезой эквивалентности или не меньшей эффективности (НМЭ) в определенной популяции пациентов. В зависимости от имеющихся данных результатов исследований оригинального препарата в сравнении с плацебо определяется первичная конечная точка и граница не меньшей эквивалентности (как правило, это половина от нижней границы 95% доверительного интервала для разницы эффекта оригинального препарата в сравнении с плацебо). Если таковых результатов нет и определить границу НМЭ невозможно, то следует использовать гипотезу эквивалентности с включением дополнительной группы плацебо в исследование.
Что означают контролируемые и неконтролируемые клинические исследования?
Контролируемые клинические исследования - это сравнительные исследования, неконтролируемые - не сравнительные. Большую значимость для решения в пользу регистрации препарата имеют контролируемые исследования, так как регулятору необходимо показать либо преимущество, либо неменьшую эффективность в сравнении с контролем.
Если исследование биоэквивалентности проведено полностью или частично в одной из стран ICH до 1 января 2016 года и препарат зарегистрирован в одной из стран ICH с использованием данного исследования, можно ли результаты исследования биоэквивалентности использовать при подаче на новую регистрацию в ЕАЭС?
Согласно Правилам регистрации использовать результаты такого исследования возможно, но обязательно это все нужно подкрепить доказательствами и обоснованиями.
При приведении досье в соответствие с требованиями ЕАЭС для дженериков от иностранного производителя можно ли подавать в модуле 5 (отчеты о клинических исследованиях) исследования биоэквивалентности, проведенные за территорией стран ЕАЭС?
Можно, но в дополнение к локальному клиническому исследованию.